
Regulatory Guide: Importing Medical Devices from China to US/EU
- Overview: Why regulatory clarity matters when working with china medical device manufacturers
- Key regulatory jurisdictions and who enforces them
- Device classification: first step with china medical device manufacturers
- Quality system expectations: ISO 13485 and FDA QSR
- Pre-market routes: clearing the US market
- Pre-market routes: meeting EU MDR requirements
- Labeling and UDI requirements for imports
- Documentation and technical file checklist for imported devices
- Customs, import entry, and roles of US Agent and EU Authorized Representative
- Testing, inspections, and supplier audits
- Post-market surveillance and vigilance obligations
- Risk-based market entry checklist for importers
- Comparison: US vs EU regulatory requirements
- Commercial considerations when sourcing from china medical device manufacturers
- Practical tips to reduce regulatory friction
- Common regulatory pitfalls to avoid
- When to use a regulatory consultant or Notified Body early
- Case study-style checklist: importer responsibilities
- How Longest fits into the regulatory and sourcing picture
- Longest flagship product advantages
- Final recommendations before placing an order
- FAQ: Common importer questions
- Next steps: practical onboarding checklist
Overview: Why regulatory clarity matters when working with china medical device manufacturers
Importing medical devices from China requires more than a purchase order — it requires regulatory alignment from product classification through market surveillance. Whether you are an importer, distributor, or OEM, understanding US and EU rules reduces customs delays, avoids refusals, and protects patients and Longest.
Key regulatory jurisdictions and who enforces them
US and EU rules dominate most medical device trade. In the US, the Food and Drug Administration (FDA) Center for Devices and Radiological Health (CDRH) enforces device pre-market and post-market requirements. In the EU, the Medical Device Regulation (MDR, Regulation (EU) 2017/745) governs device safety and performance; conformity assessment often requires a Notified Body. For sales in Great Britain, UKCA requirements apply. Understanding the responsible authority early saves time.
Device classification: first step with china medical device manufacturers
Every regulatory path starts with classification. In the US devices are classified as Class I, II, or III based on risk; many Class II devices require a 510(k) premarket notification or other premarket submissions, while Class III devices usually require a PMA. In the EU MDR, devices are classified into Class I, IIa, IIb, and III depending on intended use and inherent risk. Classification determines the evidence and conformity route required from china medical device manufacturers.
Quality system expectations: ISO 13485 and FDA QSR
Regulators expect robust quality systems. ISO 13485 is the international standard most buyers require from china medical device manufacturers; it demonstrates a consistent quality management approach. For US market entry, FDA's Quality System Regulation (21 CFR Part 820) sets requirements for design control, complaint handling, CAPA, and supplier control. Confirm that your Chinese supplier has documented procedures and evidence of implementation (audit reports, certificates, internal audit records).
Pre-market routes: clearing the US market
For the US, determine whether the device requires: 1) general controls only (often Class I), 2) a 510(k) clearance (common for Class II), 3) De Novo classification, or 4) a PMA (Class III). Importers must ensure the device is marketed under the appropriate FDA pathway and that the foreign manufacturer has registered and listed their establishment with FDA. The US Agent (designated for foreign manufacturers) is an important liaison for regulatory communications.
Pre-market routes: meeting EU MDR requirements
Under MDR, conformity assessment depends on device class. Class I devices (non-sterile, non-measuring) may allow manufacturer self-declaration, while Class IIa/IIb/III usually require Notified Body involvement for technical file review and audits. The manufacturer or their EU Authorized Representative must prepare a technical documentation package demonstrating compliance with MDR requirements, including clinical evaluation and risk management documentation.
Labeling and UDI requirements for imports
Labeling must meet both regulator expectations and marketplace requirements. The US requires English labeling and UDI placement per FDA rules for regulated devices. The EU MDR requires UDI as part of the device identification system and specific information on labeling and instructions for use. Ensure china medical device manufacturers can provide compliant labels, instructions in required languages, and barcode/UDI data in consistent formats.
Documentation and technical file checklist for imported devices
Compile a technical file or device dossier containing: device description and intended use, risk analysis (ISO 14971), clinical evaluation or justification, design and manufacturing information, labeling and IFU, verification/validation evidence, and supplier qualifications. For US imports include 510(k) or PMA documents as applicable. For EU imports ensure the Notified Body certificate (when required) and a Declaration of Conformity are available. Insist that china medical device manufacturers supply traceability records and material certificates.
Customs, import entry, and roles of US Agent and EU Authorized Representative
Importers must register shipments with border authorities and provide the regulatory documentation needed for release. In the US the importer of record works with CBP and FDA; the foreign manufacturer should have a designated US Agent. In the EU the manufacturer needs an EU Authorized Representative (if the manufacturer is outside the EU) to act as legal contact for authorities. These economic operator roles should be contractually defined with china medical device manufacturers.
Testing, inspections, and supplier audits
Do not rely solely on certificates. Conduct supplier audits, review quality records, and sample-test products before shipment. Pre-shipment inspection protocols—and third-party labs for biocompatibility, electrical safety, or sterilization validation—are essential. A documented audit trail from china medical device manufacturers reduces the risk of import alerts and elevated scrutiny.
Post-market surveillance and vigilance obligations
Both FDA and EU MDR require post-market surveillance systems. The manufacturer must collect adverse event data, perform trend analyses, and report serious incidents. Importers and distributors have obligations to forward complaints and cooperate with authorities. Confirm that china medical device manufacturers have processes for complaint handling, field corrective actions, and traceability for rapid response.
Risk-based market entry checklist for importers
Create a checklist that assigns responsibilities and timelines: classification confirmation, ISO 13485 certification review, contract for US Agent/EU Rep, pre-market submission status, labeling review, UDI data capture, customs documents, pre-shipment inspection, and post-market vigilance plan. Use this checklist to manage commercial relationships with china medical device manufacturers and to pass regulatory inspections.
Comparison: US vs EU regulatory requirements
Below is a concise comparison table to guide decisions when sourcing from china medical device manufacturers.
| Topic | United States (FDA) | European Union (MDR) |
|---|---|---|
| Primary regulator | FDA CDRH | National Competent Authorities / Notified Bodies |
| Classification | Class I / II / III (risk-based) | Class I / IIa / IIb / III (risk-based) |
| Pre-market submissions | 510(k), De Novo, PMA, or exempt | Conformity assessment; Notified Body involvement for most IIa/IIb/III |
| Quality system | 21 CFR Part 820 (QSR); ISO 13485 recommended | ISO 13485 aligned; MDR requirements for QMS and technical documentation |
| Labeling & UDI | UDI required; labeling in English | UDI required under MDR; labeling in applicable EU languages |
| Local representative | US Agent for foreign manufacturers | EU Authorized Representative for non-EU manufacturers |
| Post-market | MDR adverse event reporting, Corrections & Removals | Vigilance system, periodic safety update reports, PMS plans |
Commercial considerations when sourcing from china medical device manufacturers
Beyond regulatory fit, evaluate commercial factors: lead time reliability, minimum order quantities, spare parts availability, warranty and service arrangements, IP protections, and inventory buffers. Ensure contract terms cover regulatory non-conformances and specify responsibility for recall costs and corrective actions. Clarity on these points helps maintain supply continuity and reduces commercial risk.
Practical tips to reduce regulatory friction
Use these practical actions when working with china medical device manufacturers: 1) request full technical files early; 2) verify ISO 13485 and certificates with issuing bodies; 3) secure a documented US Agent or EU Rep; 4) perform on-site or third-party audits; 5) require UDI data files and barcode samples; 6) define complaint handling and CAPA communication protocols; 7) run pre-shipment functional tests and labeling checks. These steps prevent unexpected import holds.
Common regulatory pitfalls to avoid
Frequent issues include mismatched device classification, incomplete technical documentation, non-compliant labeling, missing US Agent/EU Rep, inadequate supplier controls, and weak traceability. Early alignment on the regulatory pathway and contractual obligations with china medical device manufacturers avoids these pitfalls.
When to use a regulatory consultant or Notified Body early
Engage regulatory experts when classification is unclear, clinical evidence is borderline, or new technologies are involved. For higher-risk devices, early dialogue with a Notified Body (EU) or pre-submission meetings with FDA (US) can illuminate data expectations and reduce rework. This is especially valuable when working with china medical device manufacturers who may need clarity on Western regulatory expectations.
Case study-style checklist: importer responsibilities
Use this practical checklist to confirm readiness before shipment: 1) classification confirmation; 2) pre-market clearance status; 3) QMS evidence from supplier; 4) labeling and UDI conformity; 5) device testing and certificates; 6) contractual agreements for recalls; 7) designated regulatory representatives; 8) customs documentation and HS codes; 9) post-market surveillance plan. Share the checklist with china medical device manufacturers and require documented responses.
How Longest fits into the regulatory and sourcing picture
Founded in 2000, Longest Medical is a global rehabilitation and aesthetic solutions provider focused on non-invasive technology. Longest has established capabilities across shock wave therapy, compression therapy, electrotherapy, electrostatic oscillation therapy, cryotherapy, ultrasound therapy, and active-passive trainers. For importers seeking reliable partners, Longest offers several advantages: long-standing industry experience since 2000, diversified product lines suitable for physical therapy, neurological rehabilitation, postoperative recovery, veterinary use, and medical aesthetics, and a track record delivering integrated equipment solutions. These strengths make Longest a candidate partner for buyers who require consistent quality and regulatory readiness when sourcing from china medical device manufacturers.
Longest flagship product advantages
Longest's product portfolio includes shockwave therapy machines (including focused shockwave systems), electrical muscle stimulation devices, air-relax compression systems, active-passive trainers, compression therapy machines, DVT medical devices, lymphatic massage and pressotherapy machines. Key advantages include non-invasive therapy focus, multi-modality offerings that enable bundled clinical solutions, and product designs oriented for clinical and rehab workflows. For importers, Longest can provide device documentation, service support, and product variants that help meet clinical needs while aligning with regulatory expectations.
Final recommendations before placing an order
Before committing to a china medical device manufacturers, verify: complete technical file availability, ISO 13485 and other certificates, willingness to provide audit access, ability to support local language labeling and UDI data, clarity on warranty/service, and agreed responsibilities for regulatory actions. Incorporate these checks into purchase orders and supplier contracts to protect your market entry and patients.
FAQ: Common importer questions
Q: Do I need an EU Authorized Representative if I import from China?
A: Yes—if the manufacturer is outside the EU, an Authorized Representative within the EU is required to act as the legal contact for authorities and to hold certain documentation under MDR.
Q: Can I import devices without ISO 13485 certification?
A: While ISO 13485 is not an absolute legal requirement in all markets, most importers and regulators expect it as evidence of a functioning QMS. Lack of ISO 13485 increases regulatory and commercial risk when sourcing from china medical device manufacturers.
Q: Who is responsible for an FDA 510(k) submission when importing?
A: The manufacturer is typically responsible for preparing the 510(k). The importer should confirm that the 510(k) exists, is valid for the device configuration, and that the foreign establishment is registered with FDA. Contractually define responsibilities for regulatory submissions and updates.
Q: What labeling languages are required for the EU?
A: Labeling and instructions must be available in the Member State language(s) where the device is placed on the market. Confirm language requirements with your EU Authorized Representative.
Q: How do I handle adverse events linked to imported devices?
A: Maintain a complaint handling and vigilance flow. Report incidents to the appropriate authority per local rules (FDA MDR reporting in the US; national competent authorities and EUDAMED obligations in the EU). Ensure china medical device manufacturers cooperate on investigations and corrective actions.
Q: Can Longest support regulatory documentation for export?
A: Longest provides product documentation and technical information for its device lines. Confirm specific regulatory support (e.g., country-specific declarations, test reports, clinical evidence) during commercial negotiations.
Next steps: practical onboarding checklist
To act now, request the following from your china medical device manufacturers: up-to-date technical file, ISO 13485 certificate, sample labeling and UDI codes, clinical data or equivalence rationale, factory audit availability, and agreed corrective action timelines. Use these documents to complete your import risk assessment and engage regulatory counsel if needed.
The electrical muscle stimulation machine Cost Guide
The Ultimate Guide to Compression Boots Recovery in Physiotherapy
Active Passive Trainer Cost in Canada: A Practical Buyer’s Guide
Shockwave vs Ultrasound Therapy: Key Differences Explained
OEM&ODM
Do customized products comply with international certification standards?
Yes. Our production processes strictly adhere to international quality management systems. Customized products can assist in applying for certifications such as CE, FDA, and TGA according to the target market's requirements, ensuring compliant sales.
2500X
Is OEM/ODM service available?
Yes. Contact us for more details. We offer professional OEM/ODM services.
RehaMoto LGT-5100D Config II
How should the device be cleaned?
The main unit should be wiped with a clean, damp cloth, and the handles can be disinfected using 75% alcohol. Straps must not be cleaned with bleach, and the cleaning temperature should not exceed 80°C.
LGT-2210DS
Is there a warranty period for the LGT - 2210DS device?
Yes, the LGT - 2210DS is covered by a warranty. For any issues, please contact our staff, and we will provide dedicated service to assist you.
LGT-2320ME
Does your product come with a warranty?
Yes, we're committed to delivering quality products and excellent after-sales service to enhance your overall experience.

Professional 12-Channel Low-Frequency Electrical Stimulation Machine LGT-2320SP
LGT-2320SP is an advanced electrical stimulation sports training station. It leads to multisite high-efficiency strength gain and realizes bilateral balanced development with motor point detection.
Specifically designed for sports medicine professionals, this device is ideal for the treatment and rehabilitation of sports-related injuries. It delivers precise electrical impulses to targeted areas of the body, promoting pain relief, muscle strengthening, and accelerated recovery.

Professional 12-Channel Low-Frequency Electrical Stimulation Machine LGT-2320BE
The LGT-2320BE is an advanced EMS machine, leveraging the power of low-frequency electrical stimulation therapy to achieve remarkable results in body sculpting.
It not only efficiently contours and tones the body but also actively promotes muscle relaxation, relieving tension and soreness. Moreover, it plays a significant role in enhancing skin elasticity, rejuvenating the skin's appearance and suppleness.
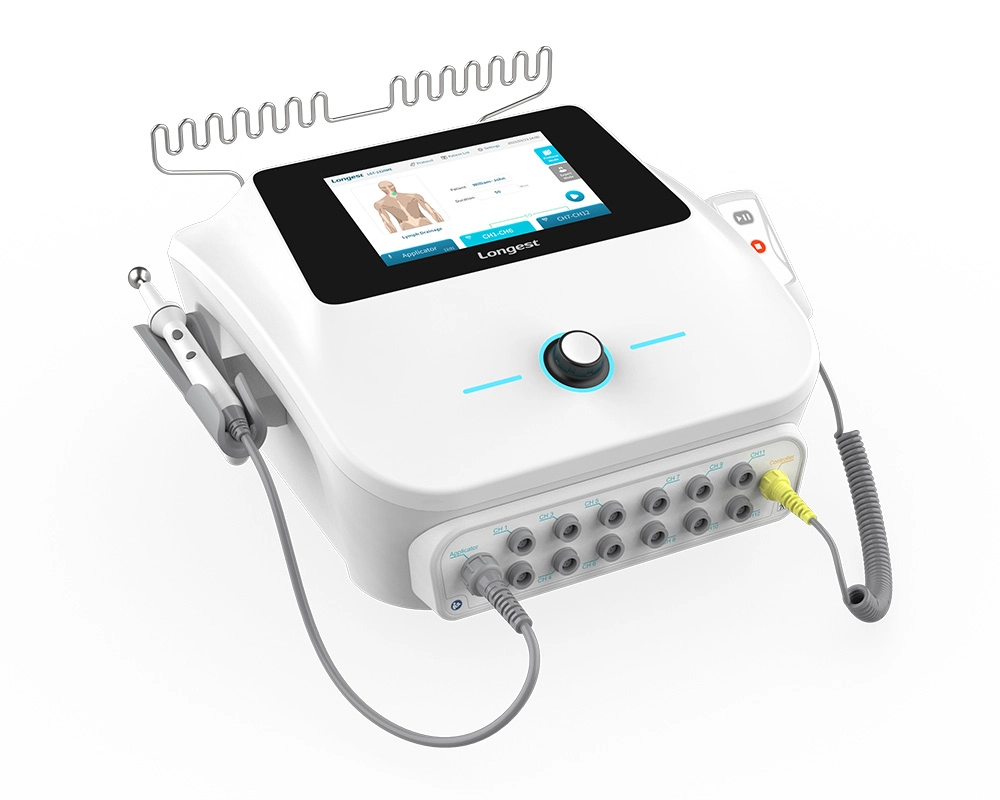
Neuromuscular Electrical Stimulation NMES Machine LGT-2320ME
The LGT-2320ME, a portable electro-stimulation therapy device, is mainly composed of the main unit, a hand switch, a pen electrode controller, and diverse types of electrodes.
It has the capacity to supply three channel groups (CH1-CH6, CH7-CH12, and the Applicator channel) with either TENS or NMES current. Specifically engineered for utilization in hospitals and clinics, this device effectively assists patients in reclaiming lost muscle strength and expedites recovery times, consequently enhancing the overall standard of patient care and bringing about more satisfactory rehabilitation outcomes.

Rehab Bike for Upper and Lower Limbs, Active-Passive Trainer (APT) RehaMoto LGT-5100D
RehaMoto controls the servo motor through the central processing unit and the biomechanical monitoring feedback system. Users can do passive, assisted, active, and constant-speed training by RehaMoto LGT-5100D Config I. The intelligent identification device realizes real - time monitoring of user training status and smooth conversion between different modes. Fully realize the best clinical training effect, and promote the recovery of users' motor function.

Call us:
+86 020 – 66353999

export@longest.cn

Address:
301 & 401 of Building 2 & Building 3, No. 96, Chuangqiang Road, Ningxi Street, Zengcheng District, Guangzhou, Guangdong Province, 511399, P.R. China.
Tips: The content on this website is a general description of the product.
The final configuration, parameters, and functions of the product may vary in accordance with the regulatory requirements of different countries and regions.
© 2025 Longest Medical. All Rights Reserved. Powered by gooeyun.
LongestMedical
LongestGloba
longest
guangzhou_longest
GzLongest