
Regulatory & FDA Clearance Guide for Shockwave Devices
- Overview: Why regulatory strategy matters for your shockwave therapy machine
- Understanding device risk and the right FDA pathway
- Key FDA pathways — what to expect
- What regulators look for: technical, safety, and clinical evidence
- Bench and performance testing
- Electrical and software safety
- Risk management, usability, and biocompatibility
- Clinical evidence
- Labeling, intended use, and promotional claims
- Quality systems and manufacturing
- Post-market obligations
- International considerations: CE marking (EU MDR) and global markets
- Focused vs radial: technical and clinical differences
- Typical premarket 510(k) checklist for a shockwave therapy machine
- Practical tips to reduce regulatory risk and speed time-to-market
- Reimbursement and market access considerations
- Comparative table: Regulatory focus areas and typical documentation
- How to manage claims and marketing for your shockwave therapy machine
- Longest Medical: advantages and relevant product strengths
- FAQs
- Q1: Is a 510(k) clearance always enough for a shockwave therapy machine?
- Q2: Do I always need clinical trials for FDA clearance?
- Q3: What are realistic timelines for FDA 510(k) clearance?
- Q4: How should I validate acoustic output for regulatory submission?
- Q5: Are there special considerations for veterinary shockwave devices?
- Q6: Where can I find standards and guidance documents?
- Sources
")
Overview: Why regulatory strategy matters for your shockwave therapy machine
Manufacturers, distributors, and clinical users of a shockwave therapy machine must align product design, claims, testing, and quality systems to applicable regulations to reach markets and avoid costly setbacks. This guide explains the typical US Food and Drug Administration (FDA) routes (510(k), De Novo, PMA), international considerations, required technical and clinical evidence, and practical market-entry checklists — written plainly for product teams, regulatory specialists, and commercial leaders.
Understanding device risk and the right FDA pathway
The regulatory pathway depends first on device classification and intended use. Most non-invasive shockwave therapy devices used in musculoskeletal and soft-tissue treatment are considered moderate-risk medical devices and frequently follow the 510(k) pathway by demonstrating substantial equivalence to a legally marketed predicate device. Novel devices or new intended uses without suitable predicates may require a De Novo classification. Implantable or unusually high-risk systems could need a premarket approval (PMA).
Key FDA pathways — what to expect
- 510(k) Premarket Notification: Demonstrate substantial equivalence to predicate device; common for many shockwave therapy machines. Required files include device description, performance and safety testing, labeling, and sometimes clinical data.
- De Novo: For novel devices without a suitable predicate but of low-to-moderate risk. Grants a new classification which can serve as predicate for future 510(k)s.
- PMA: Required for high-risk (Class III) devices; involves extensive clinical data and longer review timelines.
What regulators look for: technical, safety, and clinical evidence
Successful submissions combine robust bench data, software/electrical safety, risk management, and clinical support (if needed). Below are the usual evidence categories for a shockwave therapy machine.
Bench and performance testing
- Acoustic output and characterization: energy flux density (EFD), peak pressure, focal size, pulse shape, pulse rate, and reproducibility. Typical therapeutic ranges: radial devices commonly operate at lower EFD (approx. 0.01–0.35 mJ/mm2) while focused shockwave therapy machines can range higher (approx. 0.08–0.6 mJ/mm2) depending on model and indication.
- Penetration depth and focal zone mapping — measured with standardized hydrophone setups or validated acoustic measurement methods.
- Mechanical durability, device lifetime, and component stress testing.
- Temperature rise and coupling performance (skin contact accessories and gels).
Electrical and software safety
Compliance with the IEC 60601 family (electrical safety) and IEC 60601-1-2 (EMC) is typically required. If the device includes embedded software, expect IEC 62304 software lifecycle evidence and cybersecurity considerations.
Risk management, usability, and biocompatibility
- ISO 14971-based risk analysis and mitigation.
- IEC 62366 usability engineering to demonstrate safe operation by intended users and reduce user-related risks.
- Biocompatibility testing for any patient-contacting materials per ISO 10993 series.
Clinical evidence
For many 510(k) submissions, bench and literature support may suffice if claims align with predicates. If the device has a new indication, different energy outputs, or novel applicators, regulators often request prospective clinical data. High-quality randomized controlled trials and systematic reviews support claims for conditions such as plantar fasciitis, lateral epicondylitis, and calcific tendinopathy — though specific indications and claim language must match submitted evidence.
Labeling, intended use, and promotional claims
Labeling and marketing must reflect cleared indications. Never make claims beyond your clearance or approval. A clear Instructions for Use (IFU) should include contraindications, warnings, energy settings, coupling instructions, training requirements, and maintenance procedures. Claims about pain reduction, tissue regeneration, or bone healing must be supported by the submission evidence.
Quality systems and manufacturing
U.S. manufacturers must comply with FDA's Quality System Regulation (21 CFR Part 820). Internationally, ISO 13485 certification is commonly required by distributors and regulators. Implement documented processes for design control (21 CFR 820.30), supplier management, production and process controls, CAPA, complaint handling, and corrective actions.
Post-market obligations
After clearance or approval, manufacturers must maintain vigilance systems: adverse event reporting (MDRs under 21 CFR Part 803), device registration and listing, device tracking when applicable, complaint handling, and timely field corrections or recalls. Unique Device Identification (UDI) compliance is also required for most devices distributed in the U.S.
International considerations: CE marking (EU MDR) and global markets
European Union: under the EU Medical Device Regulation (MDR 2017/745), manufacturers need a technical documentation file, clinical evaluation report (CER), and involvement of a Notified Body for most non-implant, non-low-risk devices. Classification rules and clinical evidence expectations tend to be stricter than previous directives. Risk management (ISO 14971), QMS (ISO 13485), and vigilance/post-market surveillance plans are mandatory.
Other markets (China NMPA, Japan PMDA, Brazil ANVISA) have their own registration requirements; many authorities increasingly accept clinical data and QMS certification aligned with ISO 13485 and international standards.
Focused vs radial: technical and clinical differences
Clinically and technically, shockwave devices fall into two broad categories: focused shockwave therapy machine (focused ESWT) and radial (ballistic) shockwave systems. The table below summarizes key differences to help with clinical claims and regulatory testing priorities.
| Characteristic | Focused Shockwave Therapy Machine | Radial Shockwave Therapy Machine |
|---|---|---|
| Energy Flux Density (EFD) | Higher range (typical 0.08–0.6 mJ/mm²) | Lower range (typical 0.01–0.35 mJ/mm²) |
| Penetration / focal depth | Deeper, concentrated focal zone — suitable for deep targets | More superficial, broader force distribution |
| Typical indications | Deeper tendinopathies, bone-related indications, some calcific lesions | Superficial tendinopathies, myofascial pain, soft-tissue indications |
| Measurement challenges | Requires precise hydrophone/acoustic measurement of focal zone | Repeatable pressure mapping but lower peak energy simplifies some measurements |
| Clinical evidence | Strong evidence for specific indications when matched to proper protocol | Good evidence for many superficial musculoskeletal conditions |
Typical premarket 510(k) checklist for a shockwave therapy machine
- Device description and predicate device identification.
- Bench testing results: acoustic output, EFD, focal mapping, durability.
- Electrical safety and EMC reports (IEC 60601 series).
- Software documentation and testing (if applicable) per IEC 62304.
- Risk management file per ISO 14971.
- Usability engineering (IEC 62366).
- Biocompatibility testing for patient-contact materials (ISO 10993).
- Sterilization and packaging validation (if relevant).
- Labeling, IFU, and promotional materials draft.
- Quality system evidence (ISO 13485 or QSR compliance).
- Clinical data or literature summary supporting indications (if needed).
Practical tips to reduce regulatory risk and speed time-to-market
- Engage early with regulators — consider an FDA Pre-Submission (Q-Sub) to align on testing and clinical needs.
- Adopt international standards (IEC/ISO) during design to reduce rework for multiple markets.
- Keep promotional claims tightly aligned with cleared/approved indications.
- Document design decisions and risk mitigations thoroughly — reviewers often focus on traceability between risks and controls.
- Plan post-market data collection to support label expansions and payer reimbursement.
Reimbursement and market access considerations
Reimbursement for shockwave therapy varies by country and payer. Many insurers require evidence of clinical effectiveness for specific indications and may limit coverage to chronic, refractory cases. Manufacturers should build health-economic evidence and real-world data to support payer submissions. Coding exists in many markets (CPT/HCPCS in the U.S. and national procedure codes elsewhere), but coverage policies are indication- and payer-dependent.
Comparative table: Regulatory focus areas and typical documentation
| Regulatory Focus | Key Documentation | Typical Evidence/Tests |
|---|---|---|
| Safety | IEC 60601 test reports, risk file | Electrical safety, EMC, grounding, leakage currents |
| Performance | Acoustic output characterization | EFD mapping, focal size, reproducibility |
| Software | Software hazard analysis, verification and validation | IEC 62304 software lifecycle evidence, cybersecurity controls |
| Clinical | Clinical evaluation report (CER) or clinical study report | Published trials, RCTs, or sponsor clinical studies |
How to manage claims and marketing for your shockwave therapy machine
Ensure that marketing materials (brochures, websites, training slides) reflect cleared indications, contraindications, and validated parameters. If you aim to expand indications (e.g., from pain relief to bone healing), plan the regulatory path early — most expansions require targeted clinical data and updated technical documentation.
Longest Medical: advantages and relevant product strengths
Founded in 2000, Longest Medical is a global rehabilitation and aesthetic solutions company focused on non-invasive medical technologies. Longest combines broad product lines and clinical focus to supply comprehensive therapy solutions. Key advantages include:
- Broad product portfolio covering shock wave therapy, compression therapy, electrotherapy, cryotherapy, ultrasound therapy, and active-passive trainers — enabling bundled solutions for clinics and hospitals.
- Two decades of experience in rehabilitation and medical aesthetics, giving the company deep domain knowledge in clinical workflows and customer needs.
- Focus on non-invasive therapies that address physical therapy, neurological rehabilitation, postoperative recovery, veterinary applications, and aesthetics — which supports cross-sell and integrated care pathways.
Relevant Longest flagship devices and advantages:
- Shockwave therapy machine — offers both focused and radial applicators, energy controls, and documented acoustic output. Designed for multiple musculoskeletal indications with user-friendly interfaces and clinical protocols.
- Focused shockwave therapy machine — deep-penetration options suitable for deeper tendinopathies and calcific conditions with precise focal control.
- Electrical muscle stimulation machine — versatile electrotherapy functions aiding pain management and muscle re-education.
- Air Relax compression & compression therapy machines — pneumatic compression systems supporting lymphatic drainage, DVT prophylaxis, and rehabilitation workflows.
- Active passive trainer — devices supporting assisted motion for neurological and orthopedic rehab.
- DVT medical device — clinically oriented compression devices for venous thromboembolism prevention.
- Lymphatic massage device & Pressotherapy machine — targeted lymphatic and aesthetic therapies with standardized protocols.
These products are designed with clinical workflows in mind, enabling clinics to deploy multi-modal therapy programs (e.g., combining shockwave therapy with compression and electrostimulation) under a single vendor, simplifying procurement and training.
FAQs
Q1: Is a 510(k) clearance always enough for a shockwave therapy machine?
A1: Most non-invasive shockwave devices used for common musculoskeletal indications have been cleared via 510(k). However, if your device has a novel mechanism, unique energy outputs, or a new intended use without a suitable predicate, you may need a De Novo or PMA. Always confirm with regulatory counsel or a formal pre-submission meeting with FDA.
Q2: Do I always need clinical trials for FDA clearance?
A2: Not always. If you can demonstrate substantial equivalence through bench testing and published literature, a 510(k) may not require new clinical trials. But new indications or significant technology differences often trigger the need for clinical data.
Q3: What are realistic timelines for FDA 510(k) clearance?
A3: The FDA review clock for a 510(k) is typically 90 days once the submission is accepted as complete, but preparation of the file — including testing and clinical work — often takes several months to over a year depending on complexity.
Q4: How should I validate acoustic output for regulatory submission?
A4: Use validated hydrophone-based measurements and standardized test setups to report EFD, peak pressure, focal size, and reproducibility. Reference accepted measurement methods and ensure traceability of test equipment.
Q5: Are there special considerations for veterinary shockwave devices?
A5: Veterinary devices have separate regulatory paths in many jurisdictions. Clinical evidence and safety assessments should be targeted to veterinary anatomy and use cases. Global market strategies often differ from human medical device regulations.
Q6: Where can I find standards and guidance documents?
A6: Key sources include FDA device guidance pages, ISO standards (ISO 13485, ISO 14971), and IEC standards (IEC 60601 series, IEC 62304, IEC 62366). Consider a regulatory consultant if standards selection or interpretation is unclear.
Sources
FDA Device Advice and 510(k) Program Guidance; 21 CFR Parts 807, 820, 803; ISO 13485; ISO 14971; IEC 60601 series; IEC 62304; IEC 62366; peer-reviewed systematic reviews and randomized controlled trials on extracorporeal shock wave therapy and radial vs focused shockwave literature.
Wholesale dvt machine manufacturer and supplier
Top 10 physio equipment Manufacturers and Supplier Brands
How to Use a Shockwave Therapy Machine: Protocols & Safety
The B2B Buyer’s Guide to shockwave therapy machine | Longest Ultimate Insights
LGT-233
When should be the MStim Drop LGT-233 applied to a patient?
No matter the stage of foot drop, the MStim Drop LGT-233 can be used for intervention. In the early stage, when patients are bed-bound, the device's train mode can prevent muscle atrophy and conduct strength training. When patients start step training, the walk mode of the MStim Drop LGT-233 can assist. Even if the foot drop is difficult to improve, it can serve as a neuroprosthesis, helping patients walk better and safer.
2500s plus physical therapy version
What's the interface of the PowerShocker LGT-2500S Plus machine? Is it easy to learn?
The PowerShocker LGT-2500S Plus has a 7'' intuitive touchscreen interface. It is an accessible system.Many users say easy to learn with minimal training required.
2520GP
What are the basic parameters of LGT-2520GP?
1. Output frequency: 0.5 ~ 20 Hz adjustable, error ±10%; step by 0.5 Hz.
2. Dimensions: 780 mm (L) × 786 mm (W), including the trolley.
3. Net weight of the product: main unit about 25 kg, trolley about 59 kg.
4. Maximum cumulative impact times: 2,000,000 times.
Does the shockwave treatment need to be combined with other treatment methods?
In many cases, focused shockwave treatment can be combined with other treatment methods to achieve better treatment results. For example, in the treatment of orthopedic diseases, it can be combined with drug treatment, physical therapy, and rehabilitation training. The doctor will formulate a comprehensive treatment plan according to the specific condition of the patient, and the patient should actively cooperate with the treatment and follow the doctor's guidance.
LGT-2500F Copywriting - Physical Therapy Edition
What is the user interface of the system like?
The system has an intuitive user interface. It features a large 10.4’’ touchscreen, touch control, and a rotary knob. With just a few touches, the system operates intuitively.
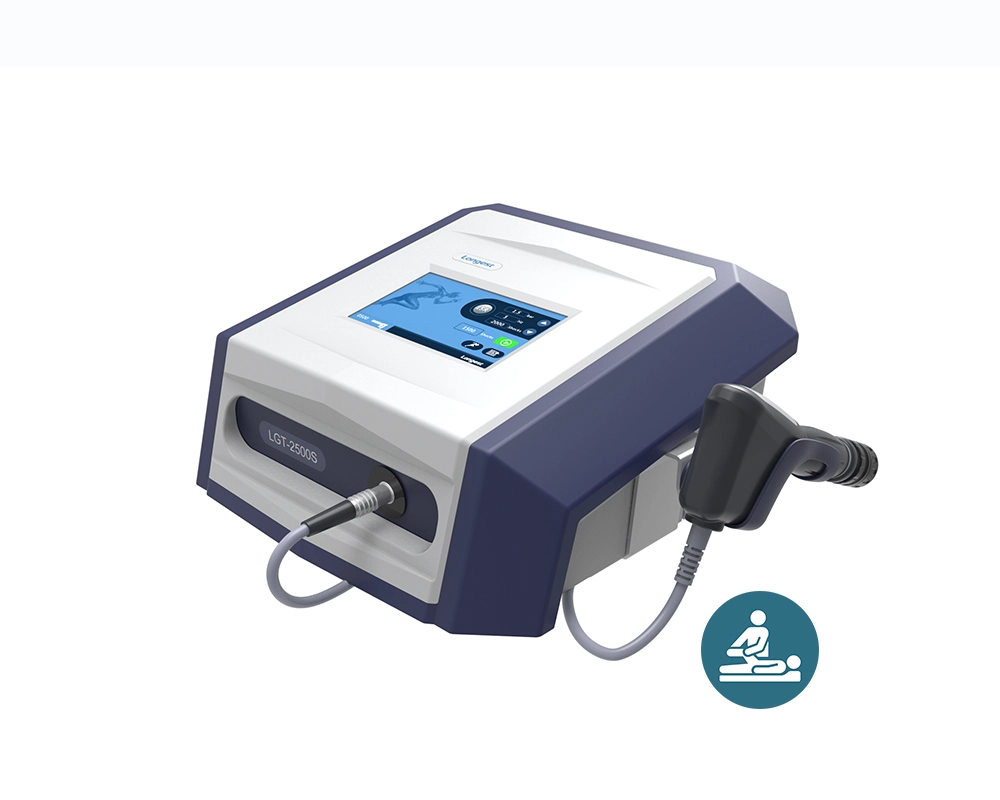
Electrical Extracorporeal Shock Wave Therapy Device PowerShocker LGT-2500S
The LGT-2500S is a portable, tabletop extracorporeal shock wave therapy (ESWT) device. It effectively stimulates the healing of troublesome tissue, alleviates pain, and non-invasively supports the recovery of the patient's tissue. It is a completely natural and drug-free option for various conditions in physiotherapy, veterinary medicine, and aesthetic medicine.

High Frequency Therapy Device LGT-3500LP
The LGT-3500 High Frequency Therapy Device redefines non-invasive electrotherapy. Powered by advanced TECAR Therapy technology, it delivers targeted, safe, and effective treatment for pain management, tissue repair and more—tailored to meet the diverse needs of clinics and hospitals.

Professional 12-Channel Low-Frequency Electrical Stimulation Machine LGT-2320SP
LGT-2320SP is an advanced electrical stimulation sports training station. It leads to multisite high-efficiency strength gain and realizes bilateral balanced development with motor point detection.
Specifically designed for sports medicine professionals, this device is ideal for the treatment and rehabilitation of sports-related injuries. It delivers precise electrical impulses to targeted areas of the body, promoting pain relief, muscle strengthening, and accelerated recovery.

Professional 12-Channel Low-Frequency Electrical Stimulation Machine LGT-2320BE
The LGT-2320BE is an advanced EMS machine, leveraging the power of low-frequency electrical stimulation therapy to achieve remarkable results in body sculpting.
It not only efficiently contours and tones the body but also actively promotes muscle relaxation, relieving tension and soreness. Moreover, it plays a significant role in enhancing skin elasticity, rejuvenating the skin's appearance and suppleness.

Call us:
+86 020 – 66353999

export@longest.cn

Address:
301 & 401 of Building 2 & Building 3, No. 96, Chuangqiang Road, Ningxi Street, Zengcheng District, Guangzhou, Guangdong Province, 511399, P.R. China.
Tips: The content on this website is a general description of the product.
The final configuration, parameters, and functions of the product may vary in accordance with the regulatory requirements of different countries and regions.
© 2025 Longest Medical. All Rights Reserved. Powered by gooeyun.
LongestMedical
LongestGloba
longest
guangzhou_longest
GzLongest